
Essai visant à évaluer l'impact du dysfonctionnement hépatique sur l'élimination de l'encorafénib en association avec le binimétinib administré à des personnes atteintes de tumeurs solides avec mutation BRAFV600E
Essai visant à évaluer l'impact du dysfonctionnement hépatique sur l'élimination de l'encorafénib en association avec le binimétinib administré à des personnes atteintes de tumeurs solides avec mutation BRAFV600E
Essai de phase I en ouvert, multicentrique, visant à évaluer l'impact des insuffisances hépatiques modérées et sévères sur la pharmacocinétique et la tolérance de l'encorafénib en association avec le binimétinib chez des adultes atteints de tumeurs solides avec mutation BRAF V600, non résécables ou métastatiques.
Comment l'essai se déroule-t-il ?
Il s’agit d'un essai de phase 1 en ouvert incluant 12 participants.
Les participants seront affectés à l'un des trois groupes suivants :
- Groupe I : 4 participants présentant une fonction hépatique normale
- Groupe II : 4 participants présentant une insuffisance hépatique modérée
- Groupe III (*) : 4 participants présentant une insuffisance hépatique sévère
(*) Avant de traiter les participants du groupe III, les données de tolérance et de pharmacocinétique seront analysées afin de s'assurer de la sécurité et de la viabilité du traitement.
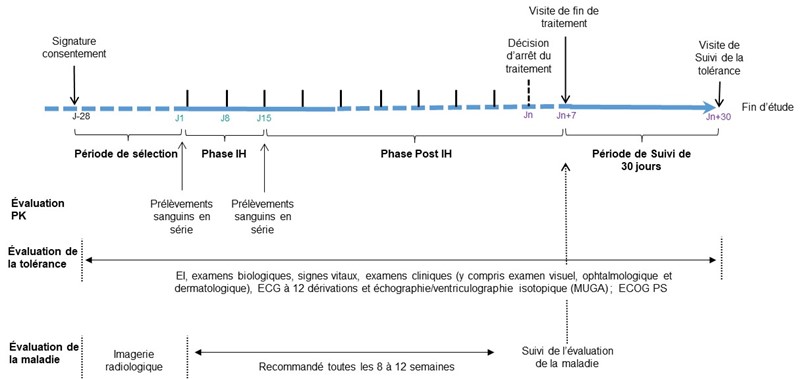
L'impact de l'insuffisance hépatique sur la pharmacocinétique sera évalué par des prélèvements sanguins en série à J1 (jour de la première administration du traitement) et à J15 (2 semaines après la première administration)
Une fois la phase d'évaluation de l'IH (2 semaines) terminée, les participants peuvent poursuivre les traitements par encorafénib et binimétinib jusqu'à ce qu'ils n'en tirent plus de bénéfices. Cela signifie que le traitement sera poursuivi tant que la maladie ne progresse pas et qu'il est bien toléré.
Après l'arrêt du traitement, les participants effectueront une visite de fin de traitement, suivie d'une visite de suivi de la tolérance (30 jours après la dernière dose de traitement).
Ce schéma synthétise les informations présentées ci-dessus :
Qui peut participer à cet essai ?
Pour participer à cet essai, les sujets doivent répondre à différents critères, parmi lesquels :
• Être un adulte atteint de tumeurs solides avec mutation BRAF V600
• Remplir les critères de fonction hépatique spécifiques à chaque groupe d'essai
Quels sont les objectifs de l’essai, et comment sont-ils évalués ?
L'objectif principal de cet essai est d'évaluer la pharmacocinétique (PK) de l'encorafénib après l'administration d'une dose unique et de plusieurs doses orales consécutives d'encorafénib en association avec le binimétinib.
Cet essai permettra également :
• d'évaluer la pharmacocinétique du binimétinib pris en association avec l'encorafénib.
• d’évaluer la tolérance en cas d’administration de plusieurs doses consécutives d’encorafénib en association avec le binimétinib en fonction du nombre et du type d'effets secondaires.
• de comparer les profils pharmacocinétiques chez des patients présentant des fonctions hépatiques altérées (modérées et sévères) et normales.
Quels sont les traitements à l’étude et comment sont-ils administrés ?
Pour chaque groupe de participants, la posologie de l'encorafénib et du binimétinib est adaptée comme suit
|
Traitement à l'étude |
Forme pharmaceutique et voie d'administration |
Dose |
Frequence |
|---|---|---|---|
|
Group I: participants présentant une fonction hépatique normale |
|||
|
Encorafenib |
6 gélules de 75 mg par voie orale |
450 mg |
1x/jour |
|
Binimetinib |
3 comprimés pelliculés de 15 mg par voie orale |
45 mg |
2x/jour |
|
Group II: participants présentant une insuffisance hépatique modérée |
|||
|
Encorafenib |
2 gélules de 75 mg par voie orale |
150 mg |
1x/jour |
|
Binimetinib |
1 comprimé pelliculé de 15 mg par voie orale |
15 mg |
2x/jour |
|
Group III: participants présentant une insuffisance hépatique sévère |
|||
|
Encorafenib |
1 gélules de 75 mg par voie orale |
75 mg |
1x/jour |
|
Binimetinib |
1 comprimé pelliculé de 15 mg par voie orale |
15 mg |
2x/jour |
aLa dose d'encorafénib pour le groupe III pourra être ajustée après analyse des données de pharmacocinétique et de tolérance des groupes I et II.