
Essai visant à démontrer que l’administration d’un comprimé de 45 mg de binimétinib est équivalente à l’administration de 3 comprimés de 15 mg
Essai visant à démontrer que l’administration d’un comprimé de 45 mg de binimétinib est équivalente à l’administration de 3 comprimés de 15 mg
Etude randomisée, monocentrique, croisée sur deux périodes, en dose unique, en ouvert pour évaluer la bioéquivalence du binimetinib administré en 3 comprimés de 15 mg et en 1 comprimé de 45 mg chez des volontaires sains
Comment l'essai se déroule-t-il ?
Il s’agit d’un essai de phase I croisé randomisé
« Randomisé » signifie que les volontaires sains seront répartis dans deux groupes au hasard :
• un groupe recevra d’abord la formulation de référence (3 comprimés de 15 mg), puis la formulation à l’étude (un comprimé de 45 mg)
• l’autre groupe recevra d’abord la formulation à l’étude, puis la formulation de référence
« Croisé » signifie que chaque volontaire sain recevra les deux formulations de manière séquentielle.
Déroulement de l’essai :
• Période de sélection avant l’administration du traitement pour s’assurer que les participants correspondent aux critères d’inclusion de l’essai.
• Première période de traitement de 5 jours. Les participants devront séjourner au centre les 3 jours suivant la première dose.
• 7 jours sans traitement, le temps que l’organisme élimine la première dose administrée.
• Deuxième période de traitement de 5 jours. Les participants devront séjourner au centre les 3 jours suivant la seconde dose.
• Une visite de fin d’étude sera réalisée 1 mois après la dernière dose, pour examen final du participant.
L’essai se déroulera dans un centre spécialisé dans les études de phase I, avec du personnel médical expérimenté et des équipements adaptés.
Ce schéma synthétise les informations présentées ci-dessus :
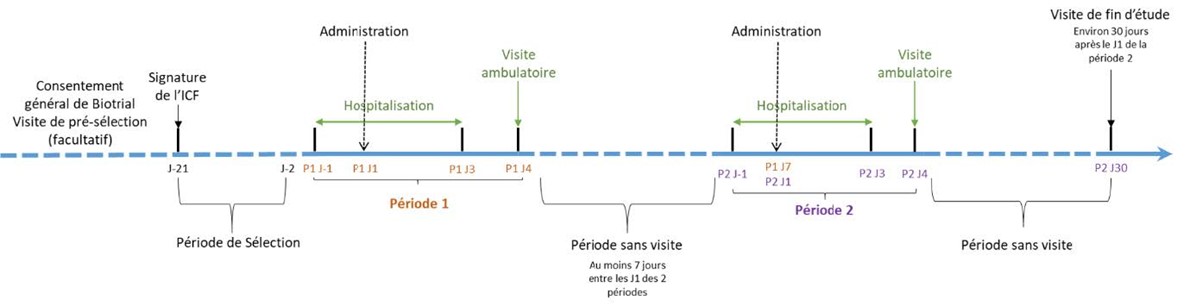
P1J1 signifie « Période 1, Jour 1 » et correspond au jour de l’administration du traitement pour la première période.
P2J1 signifie « Période 2, Jour 1 » et correspond au jour de l’administration du traitement pour la seconde période.
Qui peut participer à cet essai ?
Pour participer à cet essai, les sujets doivent répondre à différents critères, parmi lesquels :
• Avoir été identifié comme volontaire sain à l’issue des examens cliniques (analyses de sang comprises) réalisés durant la période de sélection et après étude des antécédents médicaux
• Avoir entre ≥ 18 et ≤ 65 ans, à l’exclusion des femmes enceintes et des femmes en âge de procréer
Quels sont les objectifs de l’essai, et comment sont-ils évalués ?
L’objectif principal de cet essai est de comparer la concentration de binimétinib dans le sang après administration des deux formulations.
La mesure de la concentration à différents moments après l’administration permet d’estimer :
• l’exposition totale du participant au binimétinib. L’exposition totale correspond à la quantité de médicament circulant dans le sang entre l’administration et l’élimination.
• la concentration maximale observée dans le sang
Ces paramètres pharmacocinétiques permettent d’évaluer la bioéquivalence du traitement.
Cet essai permettra également :
• De comparer d’autres paramètres pharmacocinétiques tels que le temps écoulé entre l’administration du traitement et le moment où ce dernier atteint sa concentration plasmatique maximale
• D’évaluer le profil de tolérance des deux formulations de binimétinib, en fonction du nombre et du type d’effets secondaires observés
Quels sont les traitements à l’étude et comment sont-ils administrés ?
Les participants recevront 2 doses orales uniques de 45 mg de binimétinib, avec une période d'au moins 7 jours entre les prises.
• Une administration sera effectuée à raison d’1 comprimé de 45 mg (formulation à l’étude)
• Une administration sera effectuée à raison de 3 comprimés de 15 mg (formulation de référence actuellement commercialisée)