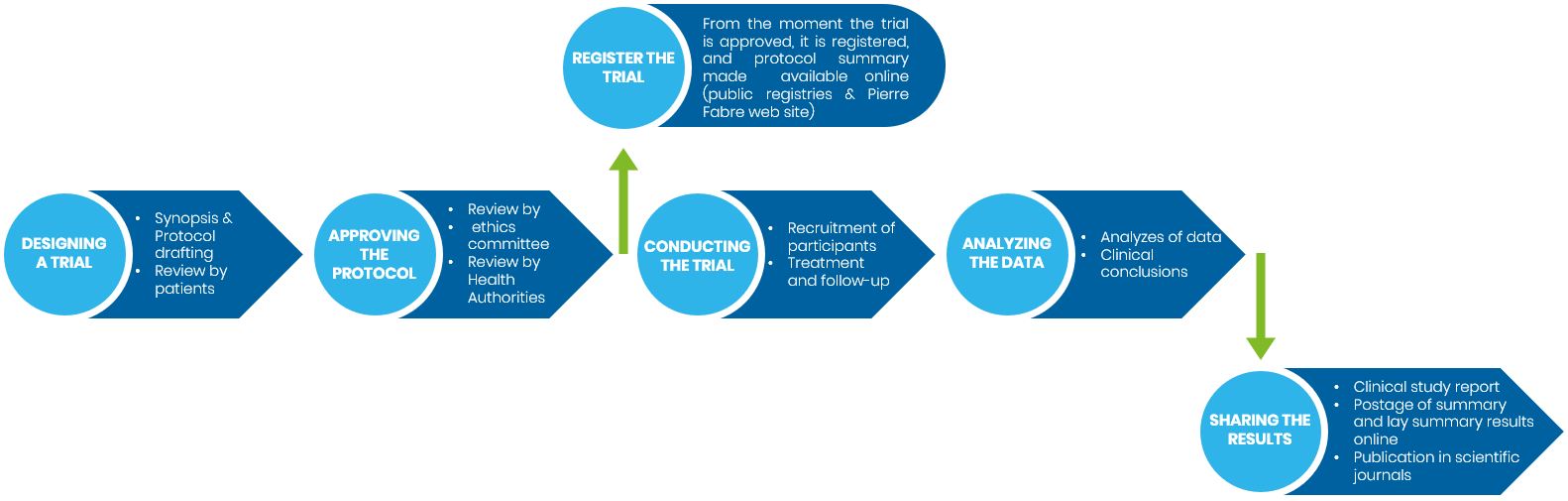
How are clinical trials designed ?
Address challenges to well-designed Clinical trials that will aptly benefit research, healthcare, and — most importantly — people is essential and implies multiple stakeholders and steps that are detailed from the design of the trial up to the reporting and disclosing of the research results.
1.
Creating/designing a trial
Creating/designing a trial
Before a clinical trial can begin, a detailed plan (named protocol) is written by the Sponsor which outlines the rationale and purpose of the trial and how it will be conducted. It includes:
- The extended name and abbreviation of the trial,
- The sponsor and collaborators,
- The study design e.g an interventional blind randomized phase III trial,
- Description of the objectives, known as primary and secondary outcomes,
- Detailed information on who may and who may not participate in the trial listed in the eligibility criteria,
- The drug or combination of drugs to be tested, the comparator(s) if any and their dosages,
- The duration of the trial,
- How side effects will be tracked, managed, and reported,
- The rules and methods that will be used to conduct it,
- How the results will be assessed and presented.
The aims of the protocol are to ensure the ethical conduct of the trial, safeguard the safety of the participants throughout the trial and assess the benefit and risk of the new drug to prevent or treat diseases.
A summary of main information contained in the protocol is also made available in both technical and easy to understand language respectively called protocol synopsis and lay protocol synopsis.
The protocol is reviewed by patient’s advocacy groups in accordance with the European clinical trial regulation to incorporate as much patient input as possible into the design of our clinical trials notably to minimize the burden of future participants
2.
Review and approval of the proposed protocol
Review and approval of the proposed protocol
An independent ethics committee must give the Sponsor permission to conduct the study, and authorization is also needed to be obtained from the health authorities in the country or countries in which the trial is to take place. This guarantee that the Sponsor follow clinical trial regulatory requirements and guidelines to protect participants safety and rights, such as Good Clinical Practice (GCP) and the protection of participant personal data, notably in Europe, under the Data Protection Regulation (“GDPR").
3.
Registration of the trial
Registration of the trial
Once the protocol is approved, the trial/study should be registered in at least one public clinical trials registry (a searchable online database, e.g., EU clinical trial register or clinicaltrials.gov) so that anyone can find and check on the details of the trial while it is ongoing, its intermediate (if any) and final results when it is terminated.
4.
Recruitment of participants and trial progress
Recruitment of participants and trial progress
Eligible patients or healthy subjects are first informed of the details of the trial as part of the informed consent process. Those who consent to participate are enrolled in the trial. Participants will have to follow the instructions and visits scheduled according to the protocol. Institutional review boards, data and safety monitoring committees, will ensure participants are protected throughout the trial.
During the trial the initial protocol can be adapted/modified by the Sponsor. This is called an amendment and is also submitted to health authorities and ethic committee for approval.
If you’re considering joining a clinical trial, visit our clinical study index page or if you want to learn more about the process of joining a clinical trial and the trial participant journey, visit our participant journey page
5.
Data analysis
Data analysis
The data come from measurements such as physical examination, blood tests, medical imaging scans like X-rays, and questionnaires completed by the participants or investigators.
After all individual participants have completed the trial, their data are anonymized, aggregated and analyzed by statistical experts on behalf of the Sponsor to obtain trial results. The study results are assessed, and conclusions drawn to determine if the study met its objectives, and whether it demonstrated that the study drug was safe and depending on the study phase, was effective.
This process can take several months, depending on the amount of data that came out from the trial.
6.
Reporting and disclosing the results to researchers, participants and general public
Reporting and disclosing the results to researchers, participants and general public
A detailed presentation of research processes and findings that includes tables and graphs is written in a formal language following international guidelines and is called a clinical study report. This report is drafted at the end of the trial or sometimes before, at timepoints defined in the protocol, notably when primary objective is assessable notably in oncology where duration of a trial can take more than 10 years.
Subsequently, the results are written into articles that are presented at scientific meetings and published in medical journals. The articles are quite technical and can be either accessed with a fee or be freely available to the study participants and the general public.
However, increasingly, the results of clinical studies are being made available to non-medical audiences. Plain language summaries results (PLSR) also called layperson summaries, are written in non-technical and easy to understand language, and in the language of the trial participants.
These are available with the clinical study results on public websites such as that of the EU clinical register at the same time but also downloadable here (visit our clinical study index page). Moreover, PLSRs are shared with relevant patient organizations.
Overview of a clinical trial/study process